2026年5月14日,北京大学第三医院心血管内科崔鸣教授团队与合作团队,在期刊Autophagy(《细胞自噬》)在线发表题为“PRKN-IMMT/MIC60 axis promotes myocardial ischemia-reperfusion injury via lysosomal degradation of GPX4”(PRKN-IMMT/MIC60轴通过溶酶体降解GPX4加重心肌缺血再灌注损伤)的研究论文。该研究揭示在缺氧/复氧条件下,PRKN通过靶向降解线粒体内膜蛋白IMMT,破坏线粒体嵴结构,进而触发GPX4的溶酶体降解导致心肌细胞铁死亡,最终加重心肌缺血再灌注(MIR)损伤。该发现为理解MIR损伤的病理机制提供了新视角,并提出了PRKN-IMMT轴作为潜在的治疗靶点。

论文截图
急性心肌梗死是严重威胁人类健康和生命的重大疾病,目前再灌注策略是其最有效的治疗手段,但缺血再灌注(MIR)同时可对心肌造成不可逆损伤,显著影响患者预后。线粒体稳态的维持对于心肌细胞的存活至关重要,深入阐明MIR过程中,线粒体损伤引发心肌细胞死亡的分子调控机制意义重大。
研究团队利用人诱导多能干细胞来源的心肌细胞以及心肌组织特异性基因敲除小鼠,分别从体外和体内建立MIR损伤模型。研究发现,PRKN介导线粒体内膜蛋白IMMT经泛素-蛋白酶体途径降解。IMMT是维持线粒体嵴结构和线粒体完整性的核心蛋白,其降解导致线粒体嵴结构破坏、膜电位下降及能量代谢障碍,并引发心肌细胞铁死亡。该研究揭示了PRKN在心肌细胞命运决策中的双重角色:在生理或轻度应激条件下,PRKN通过线粒体自噬维持线粒体稳态;在急性缺血再灌注条件下,PRKN过度激活,并通过降解IMMT引发心肌细胞铁死亡,加重心肌损伤。进一步机制探索表明,PRKN介导的IMMT降解间接诱导GPX4降解。IMMT的缺失促进线粒体与溶酶体互作,导致GPX4向溶酶体的转运与降解从而引发心肌细胞铁死亡。RAB7介导的线粒体-溶酶体接触在此过程中发挥关键作用。
综上所述,该研究揭示了PRKN在缺血再灌注条件下通过降解线粒体内膜蛋白IMMT促进心肌细胞铁死亡的病理机制,阐明了PRKN-IMMT-GPX4信号轴在MIR损伤中的关键作用,丰富了PRKN在线粒体质量控制及细胞命运中的功能认知。研究提示PRKN-IMMT轴可作为心肌缺血再灌注损伤的潜在治疗靶点,为改善急性心肌梗死患者的再灌注治疗预后提供了理论依据。
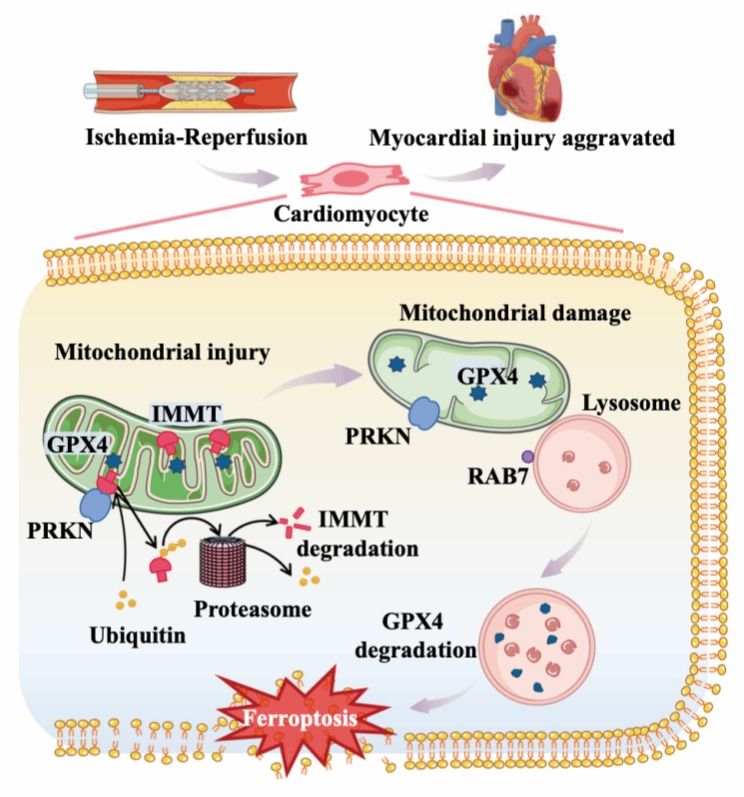
PRKN-IMMT-GPX4加重心肌缺血再灌注损伤的机制图
北京大学第三医院心血管内科博士研究生陈茜、北京大学基础医学院硕士研究生刘姝晗、博士研究生蒋梦琪为该论文共同第一作者。崔鸣、中山大学中山医学院卢广副教授和北京大学基础医学院白云教授为该论文共同通讯作者。本研究得到国家自然科学基金的资助。
论文链接:https://doi.org/10.1080/15548627.2026.2674713
作者介绍
共同第一作者
陈茜

心血管内科博士研究生
主要研究方向:心肌缺血再灌注损伤的机制研究
共同通讯作者
崔鸣

北京大学第三医院心血管内科主任、主任医师、教授、博士生导师
主要研究方向:冠心病临床诊疗及缺血再灌注损伤分子机制研究
点击了解更多